

Fototoxia, Fotoalergia, Fitofotodermatitis
13 septiembre, 2017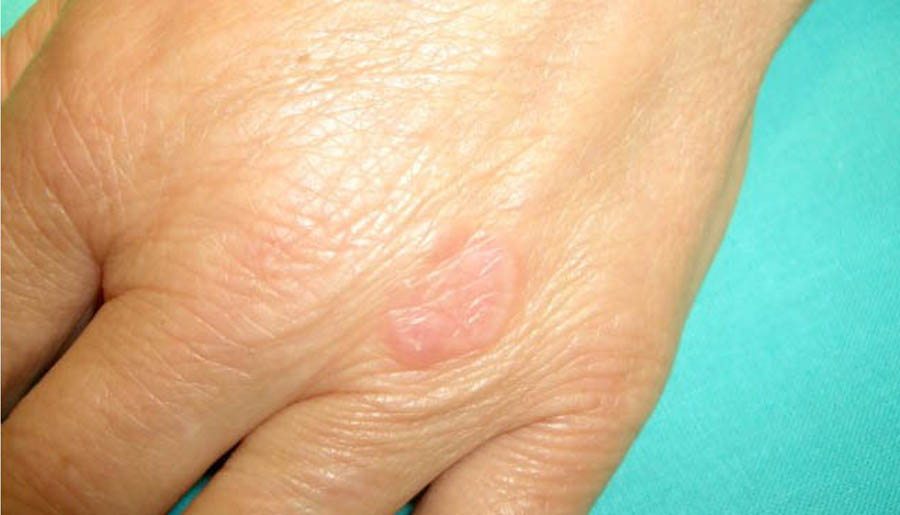
Granuloma anular
13 septiembre, 2017
1. Genodermatosis: Neurofibromatosis, Esclerosis tuberosa, Pseudoxantoma elástico, Síndrome de Ehlers-Danlos, Síndrome de Muir-Torre, Síndrome de Gorlin o del carcinoma basocelular nevoide, Síndrome de Cowden (Hamartomas múltiples), Síndrome de Gardner
Sara Alcántara Luna, Araceli Corrales Rodríguez, Ana Márquez García. Servicio de Dermatología, Hospital Universitario Virgen Macarena (Sevilla).
2. ¿Qué son?
Son dermatosis de causa genética y que frecuentemente son congénitas. Congénito es todo aquello con lo que se nace: rasgos normales o alterados (fenotipo). Puede ser genético o no.
3. ¿Qué significa genético?
Genético es todo lo que es efecto de un gen (o más). Puede ser congénito o no. Lo genético significa hereditario, no necesariamente heredable (transmisible a la descendencia). Hereditario, aunque se transmite de padres a hijos puede aparecer después del nacimiento.
3.1. Características de la herencia autosómico dominante
Las personas afectadas por una enfermedad autosómica dominante han de tener uno de los padres afectados (excepto en casos de nueva mutación); se afectan como promedio el 50% de los hijos. Sólo precisa que el gen esté en 1 cromosoma (heterocigoto).
3.2. Características de la herencia autosómico recesiva
Los padres no suelen estar afectados y se afectaría el 25% de los hijos; sólo se manifiesta si están los 2 cromosomas implicados (homocigoto). Con frecuencia son graves y es común la consanguinidad de los padres.
4. NEUROFIBROMATOSIS TIPO 1 (NF1)
4.1. ¿Qué es?
La neurofibromatosis tipo 1 es un trastorno neurocutáneo de herencia autosómica dominante, en el que en el 50% de los casos aparece de novo. Se debe a una alteración del gen de la neurofibromina, que actúa como un supresor tumoral en el control del crecimiento y la diferenciación celular. La expresividad de las manifestaciones clínicas es muy variable, incluso dentro de una misma familia, y son dependientes de la edad.
4.2. ¿Cómo se diagnostica?
Se han elaborado unos criterios para el diagnóstico clínico, que son:
- 6 o más manchas café con leche (MCCL) de diámetro mayor de 5 mm en la etapa prepuberal y mayor de 15 mm en la etapa pospuberal.
- 2 o más neurofibromas (NF) de cualquier tipo, o 1 neurofibroma plexiforme.
- Pecas axilares o inguinales.
- 2 o más nódulos de Lisch (hamartomas del iris).
- Glioma óptico.
- 1 lesión ósea característica como displasia del esfenoides o adelgazamiento de la cortical ósea de los huesos largos con/sin pseudoartrosis.
- 1 familiar en primer grado (hijo, hermano, padre o madre) con los criterios anteriores.
La presencia de 2 o más de estos criterios es diagnóstica de NF1.
Estos criterios son bastante sensibles y específicos en adultos, pero menos sensibles en niños por debajo de 8 años. Además, no tienen en cuenta otros síntomas como la dificultad en aprendizaje, los tumores malignos de la vaina nerviosa o la macrocefalia. Un estudio retrospectivo publicado en el año 2000 que recogió 1900 casos de NF1 encontró que el 46% de los casos esporádicos no cumplían criterios diagnósticos antes del primer año de vida, mientras que a los 8 años los cumplían el 97% y el 100% de los pacientes a los 20 años.
4.3. ¿Cuáles son los síntomas?
Los hallazgos dermatológicos más típicos forman parte de los criterios diagnósticos detallados anteriormente (MCCL, pecas axilares e inguinales y NF). Las MCCL aparecen a lo largo del primer año de vida en el 99% de los pacientes, y suelen aumentar en número durante la infancia. Aunque pueden existir en pacientes sanos sin NF1 asociada, se calcula que menos del 1% de los niños menores de 5 años sanos tienen más de 2 MCCL.
4.3.1. MCCL
Respetan palmas, plantas y cuero cabelludo, son de color y tamaño variable, incluso dentro de un mismo paciente. En general, tienden a aclararse a lo largo de los años y no suelen producir problemas estéticos.
4.3.2. Pecas axilares e inguinales
Clásicamente conocidas como signo de Crowe, se observan entre el tercer y el quinto año de la vida; se consideran el hallazgo cutáneo más específico. Casi el 90% de los adultos las presentan, y a menudo no sólo se limitan a los pliegues, sino que se extienden a lo largo del tronco, el cuello e incluso en torno a los labios.
4.3.3. NF
Pueden aparecer en cualquier parte del cuerpo, pero lo hacen más tardíamente, generalmente después de la pubertad. Su tamaño es variable. Un subtipo especial es el NF plexiforme superficial que suele ser congénito; los NF plexiformes superficiales pueden presentar hiperpigmentación e hipertricosis, por lo que a menudo se confunden con un nevo melanocítico congénito.
4.3.4. Otros
Máculas rojoazuladas pseudoatróficas, xantogranulomas juveniles, tumores glómicos, melanomas, nevos anémicos, hiperpigmentación generalizada, picor.
4.4. ¿Existe mayor riesgo de cáncer?
Todos los pacientes con NF1 sufren una mayor predisposición al desarrollo de neoplasias, ya sean benignas (como los NF o los tumores glómicos) o malignas. Del 8 al 12% de los pacientes con NF1 desarrollarán un tumor maligno de la vaina nerviosa periférica, generalmente en la zona de un NF plexiforme. El dolor persistente y el crecimiento acelerado deben considerarse signos de alarma, y el picor local podría indicar la presencia de un tumor del sistema nervioso central. Por otro lado, hay mayor incidencia de sarcomas, rabdomiosarcomas, neuroblastomas, tumores gastrointestinales estromales, feocromocitomas y cáncer de mama. Finalmente, los niños con NF1 tienen un riesgo entre 200 y 500 veces mayor al de la población normal de sufrir una leucemia mielomonocítica juvenil, independientemente de la concurrencia o no de xantogranulomas juveniles; sin embargo, no parece adecuado hacer despistaje rutinario salvo que los hallazgos asociados lo aconsejen (hepatoesplenomegalia, adenopatías, palidez, petequias…)
4.5. ¿Cuál es el pronóstico y tratamiento?
El curso de la enfermedad es muy variable. Deben examinarse todos los miembros de la familia.
A largo plazo los pacientes tienen una menor esperanza de vida por el desarrollo de los tumores malignos, la hipertensión asociada a la estenosis de la arteria renal o a un feocromocitoma u otras complicaciones.
El tratamiento es puramente sintomático, indicándose cirugía por motivos cosméticos o por sospecha de malignización de un neurofibroma. Las manifestaciones viscerales se corregirán según la especialidad dicte. Es importante el consejo genético, pues la mitad de los hijos de un paciente con NF-1 tendrán la enfermedad.
5. NEUROFIBROMATOSIS TIPO 2 (NF2)
5.1. ¿Qué es?
La NF2 es un síndrome de baja incidencia con herencia autosómica dominante que predispone a la aparición de tumores en el sistema nervioso pudiendo acompañarse de manifestaciones cutáneas y oculares. Se debe a mutaciones del gen que codifica la formación de la proteína denominada merlina o schwanomina que actúa como supresora de tumores.
5.2. ¿Cuáles son los síntomas?
Las manifestaciones más frecuentes se producen a nivel neurológico, ocular y cutáneo.
5.2.1. Neurológicas
La más característica es el schwanoma bilateral del VIII par craneal, presente en el 90-95% de los pacientes a los 30 años, y que es uno de los criterios diagnósticos de la enfermedad. Otras manifestaciones características incluyen schwanomas de otros pares craneales, meningiomas intracraneales, tumores espinales y tumores del sistema nervioso periférico.
5.2.2. Oculares
Destacan las cataratas (60-81%), las membranas epirretinianas (12-40%) y los hamartomas retinianos (6-22%).
5.2.3. Cutáneas
Tienen una importancia menor en la NF2 si las comparamos con las de la NF1 donde son un importante criterio diagnóstico. Entre el 59 y el 68% de los pacientes con NF2 las presentan, sin embargo, solamente el 10% tendrán más de 10 lesiones. Los pacientes con enfermedad severa presentan con mayor frecuencia lesiones cutáneas y estas son más numerosas. Aunque un 33% de los pacientes pueden presentar MCCL, aparecen normalmente en número menor de 6 a diferencia de lo que sucede en NF1. La morfología de las lesiones cutáneas es muy variada; las más frecuentes son placas tumorales hiperpigmentadas que pueden asociar un aumento de pelo en su superficie. En segundo lugar nos encontramos con nódulos subcutáneos esféricos generalmente localizados sobre nervios periféricos y dolorosos a la palpación.
6. NEUROFIBROMATOSIS TIPO 5 (SEGMENTARIA)
6.1. ¿Qué es?
Se caracteriza por la afectación de un segmento del cuerpo y la ausencia de antecedentes familiares de la enfermedad. Puede haber MCCL aisladas, NF aislados, MCCL y neurofibromas simultáneamente o NF plexiformes aislados.
7. ESCLEROSIS TUBEROSA (ET)
7.1. ¿Qué es?
Es una genodermatosis de herencia autosómica dominante en un 30%, siendo el resto de los casos nuevas mutaciones. Se caracteriza por la aparición de hamartomas en múltiples órganos, incluyendo la piel y el sistema nervioso central. Su patogénesis se fundamenta en la alteración de las proteínas hamartina o tuberina.
7.2. ¿Cómo se diagnostica?
El diagnóstico de la ET es fundamentalmente clínico y se basa en los siguientes criterios:
7.2.1. Criterios mayores:
- Angiofibromas o placa fibrosa de la frente
- Fibromas periungueales no traumáticos
- Máculas hipomelanóticas (3 o más)
- Placa chagrin (nevus del tejido conectivo)
- Hamartomas retinianos múltiples
- Tuberosidades corticales
- Nódulos subependimarios
- Astrocitoma subependimario
- Rabdomioma cardíaco, único o múltiple
- Linfangioleiomatosis
- Angiomiolipoma renal
7.2.2. Criterios menores:
- Depresiones dentales múltiples
- Hamartoma rectal polipoideo
- Quistes óseos
- Alteraciones en la migración de la sustancia blanca
- Fibromas gingivales
- Hamartomas no renales
- Manchas acrómicas en la retina
- Hipopigmentación en confeti
- Quistes renales múltiples
Para realizar el diagnóstico se requieren dos criterios mayores o uno mayor y dos menores;
Diagnóstico probable: uno mayor y otro menor
Diagnóstico posible: uno mayor y dos o más menores.
7.3. ¿Cuáles son los síntomas?
La clínica típica consiste en la triada clásica de múltiples angiofibromas, epilepsia y retraso mental, aunque esta asociación sólo es evidente en el 29% de los pacientes.
Los primeros síntomas de la enfermedad suelen ser los neurológicos. Pueden presentarse convulsiones focales o generalizadas a los pocos meses de vida, alteraciones neurológicas focales y retraso en el desarrollo psicomotor que puede ser progresivo y severo. A partir de los 8-10 años puede iniciarse la insuficiencia renal y la insuficiencia cardíaca. También pueden detectarse facomas retinianos (placas amarillentas o grisaceas), rabdomiomas cardiacos (responsables de la insuficiencia cardíaca), tumores renales (angiolipomas y quistes responsables de la insuficiencia renal), hepáticos, testiculares y del aparato gastrointestinal y quistes pleurales, pulmonares y óseos.
Las lesiones cutáneas son uno de los principales marcadores de la enfermedad. Las primeras en observarse son las máculas hipomelanóticas en «hoja de fresno», mucho más visibles al examen con luz de Wood y presentes en el 80% de los niños afectos, que pueden apreciarse a los pocos meses de edad. No es infrecuente que se asocie a un mechón de cabello blanco o poliosis. A pesar de la gran variabilidad clínica de la enfermedad y su penetrancia variable, los angiofibromas faciales se encuentran presentes en el 83-90% de los casos. Se localizan de forma preferente en surco nasogeniano, mejillas y mentón; en cuanto al número y el tamaño de los mismos no existe correlación entre la intensidad de estas lesiones y el daño del sistema nervioso central. Suelen aparecer a lo largo de la primera década de la vida y se estabilizan en la adolescencia, persistiendo de por vida. Otra lesión muy característica es la denominada placa en chagrín, que suele aparecer después de los 5 años de edad. Se trata de una lesión de varios cm de diámetro, situada por lo general a nivel dorsal o lumbar, del color de la piel y tacto como de cuero tratado. Se trata en realidad de un nevo del tejido conectivo, caracterizado por una fibrosis dermohipodérmica. También son característicos los fibromas peri y subungueales, conocidos como tumores de Koenen, de consistencia elástica, rosados o del color de la piel, de superficie lisa, que aparecen habitualmente a partir del borde proximal de la uña y con frecuencia producen una hendidura longitudinal de la misma. Tienden a surgir tras la pubertad.
7.4. ¿Cuál es el pronóstico y tratamiento?
La principal causa de morbimortalidad de la ET se debe a las manifestaciones neurológicas.
Los casos graves tienen una mortalidad del 30% antes de los 5 años y del 70% antes de la vida adulta.
En el tratamiento de los angiofibromas se han empleado diversas opciones terapeúticas, incluyendo escisión simple, criocirugía, curetaje, dermoabrasión, láser de CO2 y terapia fotodinámica, entre otras. Ningún tratamiento se ha mostrado efectivo para controlar su aparición y/o evitar las recidivas. La epilepsia incontrolada puede requerir intervención quirúrgica. El tratamiento de las lesiones viscerales es poco satisfactorio.
8. PSEUDOXANTOMA ELÁSTICO (PXE)
8.1. ¿Qué es?
Es una enfermedad del tejido conectivo hereditaria, caracterizada por una fragmentación y calcificación progresiva de las fibras elásticas de la piel, paredes vasculares (sistema cardiovascular) y ojo. Se han descritos varias formas clínicas, algunas de las cuales se heredan de forma autosómico dominante y otras autosómico recesiva; además existen casos esporádicos. El gen responsable es el ABCC6, que codifica una proteína transportadora de membrana susceptible de influenciar la composición de la matriz extracelular.
8.2. ¿Cómo se diagnostica y cuáles son los síntomas?
Se han propuesto unos criterios para establecer el diagnóstico. El síndrome completo de PXE clínicamente se caracteriza por la tríada clásica de: a) manifestaciones cutáneas (pápulas amarillentas localizadas preferentemente en áreas de flexión) b) alteraciones oculares (pigmentación moteada de la retina, estrías angioides, hemorragias retinianas) y c) manifestaciones cardiovasculares (diátesis hemorrágica, cardiopatía isquémica, claudicación intermitente, enfermedad tromboembólica).
8.2.1. Cutáneas
Consisten en pápulas amarillentas de 1 a 3 mm, lineales o en placas, reticuladas, que frecuentemente presentan telangiectasias en superficie, situadas en superficies laterales de cuello, región infraclavicular, axilas, abdomen, ingles, periné, paladar blando, cara interna de labios y mucosa intestinal. Suele iniciarse en la infancia y no suele modificarse. Se asocian a hiperlaxitud cutánea.
8.2.2. Oculares
Estrias angioides de color gris pizarra que rodean la desembocadura del nervio óptico, son bilaterales y simétricas, y se observan entre los 20 y los 40 años. Pueden dar lugar a deterioro visual (principalmente pérdida de visión central).
8.2.3. Vasculares
Son frecuentes las alteraciones arteriales, que dan lugar a claudicación intermitente, angor pectoris (incluso en niños), hemorragias gastrointestinales y hemorragia cerebral. El daño de la arteria renal puede dar lugar a una hipertensión secundaria.
Los síntomas no suelen estar presentes en el momento del nacimiento, siendo los hallazgos cutáneos los primeros en hacerse evidentes hacia la segunda década de la vida.
8.3. ¿Cuál es el tratamiento?
El aspecto más importante es asegurar que las complicaciones vasculares puedan prevenirse o actuar de forma rápida y apropiada cuando aparezcan.
9. SÍNDROME DE EHLERS-DANLOS
9.1. ¿Qué es?
Bajo este síndrome se agrupan diversas enfermedades hereditarias, algunas de herencia autosómico dominante, otras autosómico recesiva y otras ligadas a X, cuya base patogénica común es una alteración en la biosíntesis del colágeno.
9.2. ¿Cómo se diagnostica y cuáles son los síntomas?
Para muchos pacientes los síntomas son tan mínimos que su diagnóstico puede pasar desapercibido. El diagnóstico es clínico siendo la triada diagnóstica la hiperextensibilidad cutánea, hiperlaxitud articular y fragilidad del tejido conectivo.
- Signos cutáneos: la piel muestra hiperextensibilidad pero no laxitud (es decir, recupera su posición inicial sin pliegues ni arrugas cuando deja de estirarse), siendo frágil, pálida, fina con erosiones tras mínimos traumatismos. Tendencia a hematomas. La cicatrización es lenta y habitualmente con cicatrices atróficas en papel de fumar, pudiendo aparecer incluso pseudotumores moluscoides. Otros cambios cutáneos son las pápulas piezogénicas, pequeñas herniaciones de grasa a nivel de las superficies laterales de los talones.
- Otras manifestaciones frecuentes son las luxaciones y la consiguiente inestabilidad articular responsable del retraso en el inicio de la marcha en la infancia. La aparición de una pronunciada cifoescoliosis que se acentúa con la edad es responsable de insuficiencia respiratoria.
- También pueden presentar alteraciones oculares, complicaciones vasculares y obstétricas.
10. SÍNDROME DE MUIR-TORRE (SMT)
10.1. ¿Qué es?
Es una genodermatosis de transmisión autosómica dominante con penetrancia y expresividad variables.
Se caracteriza por la asociación de tumores cutáneos derivados de las glándulas sebáceas y neoplasias viscerales, destacando su carácter familiar, lenta evolución y buen pronóstico vital. Además ha sido descrita en asociación a otras enfermedades de base genética como déficit de alfa-1-antitripsina e hiperlipidemia familiar.
Forma parte del síndrome de carcinomatosis múltiple familiar o síndrome de Lynch II, relacionado con mutaciones en el gen MSH2 (algunos en MLH1), recomendándose la realización de despistaje en familiares asintomáticos, dado el riesgo de desarrollo de procesos neoplásicos en diferentes órganos.
10.2. ¿Cómo se diagnostica?
Existen criterios diagnósticos, aceptando como únicos marcadores cutáneos el adenoma, el epitelioma sebáceo o sebaceoma y el carcinoma sebáceo.
10.3. ¿Cuáles son los síntomas?
El adenoma sebáceo es el tumor más frecuente y se localiza fundamentalmente en región facial, no metastatiza y no recidiva.
El carcinoma sebáceo, originado a partir de las glándulas de Meibomio, es una neoplasia anexial con diferenciación sebácea y capacidad metastásica. La localización puede ser ocular (75%) y extraocular (25%). El Pronóstico y la asociación con el SMT no están relacionados con la localización, presentando una tasa de recurrencia local y metástasis en el 33% y el 25% de los casos respectivamente.
Las hiperplasias sebáceas se asocian frecuentemente, aunque no se consideran un marcador diagnóstico. Suelen aparecer en personas de edad avanzada, pero no implican un aumento de la incidencia de neoplasias viscerales malignas.
Los queratoacantomas con diferenciación sebácea aparecen descritos en un 20% de los pacientes, mostrando predilección por la edad media y localizándose en áreas fotoexpuestas.
En este amplio espectro de proliferaciones sebáceas presentes en el SMT se incluyen, además, el hamartoma quístico foliculosebáceo, el epitelioma basocelular con diferenciación sebácea, el tricofoliculoma sebáceo y el sebomatricoma.
El número de tumores viscerales diagnosticados en cada paciente es variable y oscilan entre 1 y 9, aunque suelen ser únicos. La relación temporal entre la aparición de las lesiones cutáneas y el diagnóstico del cuadro neoplásico visceral es variable.
El carcinoma colorrectal es el tumor asociado con mayor frecuencia (aproximadamente el 60% del total). Presenta algunas particularidades, tales como su inicio en edades tempranas y su localización proximal al ángulo esplénico, a diferencia de la población normal, en la que el colon distal y el recto son las localizaciones más comúnmente afectas. El 28% de los pacientes desarrollan, además, poliposis colónica, asociación que constituye un marcador de riesgo para la aparición de otras neoplasias digestivas.
En el 20%-25% de los casos aparecen tumores genitourinarios en todas sus localizaciones. Los órganos implicados con mayor frecuencia son útero, riñón, ovario y próstata. Menos habitual es la presencia de carcinoma de mama y procesos hematológicos, como linfomas Hodgkin y no Hodgkin, leucemia linfática crónica y policitemia vera. Excepcionalmente, el SMT se desarrolla en el contexto de tumoraciones de cabeza y cuello, intestino delgado, estómago, páncreas o melanoma.
La existencia de múltiples familiares afectos de neoplasias internas en un paciente afecto de SMT es frecuente, considerándose éste una expresión fenotípica más completa del síndrome de Lynch.
Es importante remarcar la conveniencia de la realización de un estudio sistémico completo y un seguimiento posterior a todo paciente diagnosticado de una o varias tumoraciones cutáneas de origen sebáceo dada su posible asociación a neoplasias viscerales internas. Al tratarse de un cuadro genético debe estudiarse la mayor cantidad posible de familiares de pacientes afectos, incluso asintomáticos, ya que en ellos el riesgo de desarrollar neoplasias internas malignas es mayor que en el resto de la población.
10.4. ¿Cómo se debe proceder ante la sospecha de SMT?
El SMT es una genodermatosis autosómica dominante, que viene definido por la presencia en un paciente de un tumor cutáneo de naturaleza sebácea (Adenoma sebáceo, Sebaceoma, Epitelioma sebáceo, Carcinoma sebáceo, Carcinoma Basocelular con diferenciación sebácea, Queratoacantoma con diferenciación sebácea) y una neoplasia visceral maligna (Colorrectal y genitourinario principalmente) ó la presencia simultánea de queratoacantomas múltiples, tumores malignos viscerales múltiples e historia familiar de SMT. Si se cumplieran una de las dos premisas estaría indicado el análisis de la inestabilidad de microsatélites (MSH-2, MLH-1, MSH-6), ya que si el resultado del estudio genético fuera positivo serían necesarias revisiones periódicas por los servicios de digestivo, ginecología y urología.
10.5. ¿Cuál es el pronóstico?
Las neoplasias suelen ser múltiples y de comienzo precoz, pero el pronóstico es bastante bueno y la incidencia de metástasis baja.
11. SÍNDROME DE GORLIN O DEL CARCINOMA BASOCELULAR NEVOIDE
11.1. ¿Qué es?
Es un trastorno hereditario autosómico dominante, con penetrancia elevada y expresión fenotípica variable, caracterizado por alteraciones del desarrollo y una predisposición a diferentes tipos de tumores. Se debe a mutaciones en un gen supresor de tumores (PTCH1). Sin embargo, entre el 33 y el 60% de los pacientes con esta enfermedad no presentan historia familiar y se cree que son producto de mutaciones surgidas de novo.
11.2. ¿Cómo se diagnostica y cuáles son los síntomas?
Las manifestaciones clínicas incluyen carcinomas basocelulares, queratoquistes odontogénicos mandibulares, depresiones palmoplantares, también conocidas con el término inglés de “pits” y calcificación ectópica de la hoz cerebral, que son consideradas criterios mayores para el diagnóstico. Otras manifestaciones menos frecuentes o menos específicas (consideradas criterios menores) son dismorfismo facial (macrocefalia, paladar hendido, frente abombada, hipertelorismo moderado o severo), anomalías vertebrales y aumento de la frecuencia de otros tumores, como fibromas ováricos y meduloblastomas.
11.3. ¿Cuál es el pronóstico?
El meduloblastoma se ha descrito en un 5% de los pacientes, pudiendo ser la forma de inicio de la enfermedad y siendo lo que marca el pronóstico de la enfermedad.
11.4. ¿Cuál es el tratamiento?
La extirpación quirúrgica de todas las lesiones tumorales, aunque deseable, a menudo es imposible por la gran cantidad de lesiones presentes. No se aconseja radioterapia pues puede inducir mayor número de lesiones. La terapia fotodinámica (aplicación de fotosensibilizantes e irradiación con luz visible) es útil para el tratamiento de carcinomas basocelulares. También el tratamiento con imiquimod puede ser útil para controlar las proliferaciones basaloides.
12. SÍNDROME DE COWDEN (HAMARTOMAS MÚLTIPLES)
12.1. ¿Qué es?
Es una genodermatosis de herencia autosómica dominante, que se caracteriza por la presencia de múltiples tumores cutaneomucosos (tricolemomas faciales) y viscerales, junto con tendencia al desarrollo de carcinoma mamario y tiroideo. Genéticamente se ha establecido un defecto en el gen supresor de tumores PTEN.
12.2. ¿Cuáles son los síntomas?
El signo de presentación en la infancia puede ser una macrocefalia. La edad media de comienzo de las lesiones cutáneas son los 22 años. Los tricolemomas se presentan como pápulas verrugosas de color piel o amarilla-parduscas con hiperqueratosis superficial, generalmente agrupadas alrededor de la boca, nariz y ojos. En palmas y plantas se pueden observar hasta en la mitad de los casos queratosis punctata con depresión central. Las lesiones mucosas consisten en pápulas de color piel que pueden cubrir grandes zonas y dar un aspecto “en empedrado” característico. Con menor frecuencia se encuentran otras lesiones cutáneas como ganglioneuromas, lipomas, angiolipomas, quistes epidérmicos y cambios pigmentarios.
Además presentan con cierta frecuencia facies adenoidea, cifoescoliosis y paladar ojival. Las lesiones tumorales benignas internas son muy variadas. La patología mamaria es el proceso asociado que mayor importancia y repercusión tiene.
12.3. ¿Cuál es el pronóstico?
Tanto la patología mamaria como la tiroidea son excepcionales en el varón, por lo que en este grupo de población el pronóstico es mucho mejor.
13. SÍNDROME DE GARDNER
13.1. ¿Qué es?
Genodermatosis que se hereda con un patrón de herencia autosómico dominante. En algunos casos se han descrito mutaciones en el gen APC, ligado a la supresión tumoral.
13.2. ¿Cuáles son los síntomas?
Las lesiones cutáneas y óseas se manifiestan durante la infancia generalmente antes de la poliposis intestinal característica que suele aparecer entre los 25-35 años. Las neoplasias se desarrollan 15-20 años más tarde.
Las lesiones cutáneas consisten en quistes epidérmicos, frecuentemente localizados en cabeza y cuello, con una tendencia al aumento progresivo en número y tamaño durante un tiempo hasta estabilizarse. Pueden estar presentes desde el nacimiento.
Los osteomas, presentes hasta en un 80 % de los pacientes, se han podido constatar hasta en niños de 8 años. Afectan generalmente a los huesos de la cara, en especial al maxilar o a la mandíbula. También se pueden encontrar tumores desmoides, fibromas y, menos frecuentemente, lipomas, leiomiomas, tricoepiteliomas, neurofibromas, quistes ováricos e hiperplasia linfoide de íleon terminal.
13.3. ¿Cuál es el pronóstico?
La degeneración maligna de los pólipos intestinales aparece en el 100% de los pacientes y comienza generalmente entre los 20-30 años, aunque hay casos descritos incluso a los 9 años. Por esta razón, en ocasiones, se recomienda colectomía profiláctica.
14. Véase también
- Cicatriz, Cicatrización, Cicatriz hipertrófica, Queloide
- Criocirugía o crioterapia
- Láser y Luz intensa pulsada (IPL)
- Melanoma
- Quiste Infundibular, Quiste Tricolémico, Lipoma

